
1Research Group in Dermatology GRINDERM, Free University, Cali, Colombia.
*Corresponding author: Ana Maria Varela Baena & Lina Johanna Moreno Giraldo
Research Group in Dermatology GRINDERM, Libre University, Cali, Colombia.
Email: Anam-varelab@unilibre.edu.co &
lina.johanna.moreno@correounivalle.edu.co
Received: July 06, 2024
Accepted: August 05, 2024
Published Online: August 12, 2024
Journal: Journal of Artificial Intelligence & Robotics
Copyright: © Varela AM & Moreno LJ (2024). This Article is distributed under the terms of Creative Commons Attribution 4.0 International License.
Citation: Baena AMV, Giraldo LJM. Identification of a new and de novo heterozygous variant in the TP63 gene associated with ADULT syndrome using state-of-the-art bioinformatics and artificial intelligence. J Artif Intell Robot. 2024; 1(1): 1002.
The TP63 gene located on chromosome 3q28 [1], encodes the tumor protein p63 (TP63 MIM*603273), belonging to the p53 transcription factor family [2], which is essential in the development of ectodermal structures comprising skin, sweat glands, hair, nails and teeth. It is also involved in the development of limbs, craniofacial and other organs/tissues [1]. Its pathogenic variants have been related to 8 phenotypes, of which 7 share overlapping findings of ectodermal dysplasia, limb anomalies, cleft lip/palate [3], these include: 1. ankyloblepharon-ectodermal defects-cleft lip/palate (ACS) syndrome, 2. Rapp-Hodgkin syndrome, which some authors include in the AEC, 3. Acro-dermo-ungueal-tear tooth syndrome (ADULT), 4. Ectrodactyly, ectodermal dysplasia, cleft lip/palate syndrome (EEC3), 5. Limb-mammary syndrome, 6. Split hand/foot malformation type 4 (SHFM4) 7. Isolated cleft lip/palate (orofacial cleft) [1,3].
These phenotypes have been related to heterozygous pathogenic variants and specifically the ADULT syndrome (Acro-dermato-ungual-lacrimal-tooth), is related to variants in the protein domains, mainly the transactivator domain of the P63 protein [3], this is a rare genetic condition, with an autosomal dominant inheritance pattern described for the first time in 1993 by Propping and Zerres [4]. Characterized by the presence of ectodermal dysplasia with hypohidrosis, nail dysplasia, hair alterations (hypotrichosis), dental anomalies (conical teeth, hypodontia), in addition to syndactyly and/or ectrodactyly, mammary hypoplasia, skin lesions, ephelides, and tear duct anomalies [5,6], at birth the most striking finding may be limb anomalies such as ectrodactyly or also called split hand/foot which is visible even in prenatal ultrasound studies [7] and becomes a red flag for a syndromic ectrodactyly of genetic origin, as the patient in our case presented. However, after observation and growth of the patient, findings of ectodermal dysplasia with hypotrichosis, xerosis, nail dysplasia, conical teeth, hypodontia and obstruction of the lacrimal duct with epiphora and frequent episodes of conjunctivitis in the child becomes. This leads us to the clinical suspicion of syndromic ectrodactyly, which requires genetic confirmation given the great genotypic and phenotypic heterogeneity and the importance of a specific diagnosis, in order to establish targeted therapeutic measures, prognosis and genetic counseling, bringing us closer to precision medicine [8].
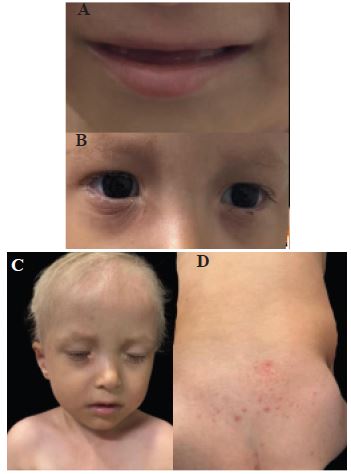
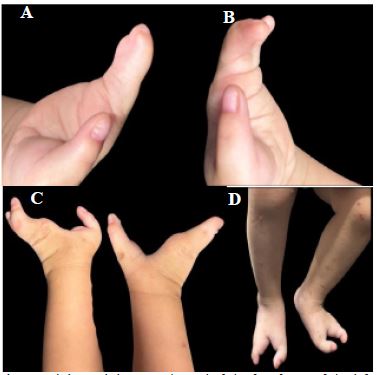
We present the case of an older male infant, 21 months old, product of a second pregnancy of non-consanguineous parents, with no family history of hereditary genetic diseases, born at 38.2 weeks without complications, with prenatal ultrasound diagnosis of ectrodactyly of the 4 extremities, at birth physical examination confirms ectrodactyly of the 4 extremities.
During infancy, the patient presented outbreaks of intermittent eczema predominantly in the cervical and antecubital folds, ungϋeal dysplasia, dental anomalies, hair anomalies, with repeated episodes of conjunctivitis and epiphora secondary to obstruction of the lacrimal duct.
Physical examination revealed: Skin: Skin phototype type III, generalized xerosis, areas of discrete thickening predominantly in folds, no ulcers, no erosions, no presence of ephelides or lentigines; Head and scalp: Presence of hypotrichosis in the scalp, eyebrows, and eyelashes with light-colored hair, blond hair, associated with depopulation in eyebrows and eyelashes with light hairs, bilateral lacrimal duct obstruction, no alterations in lip or palate, conical teeth with subsequent hypodontia. Extremities: Ectrodactyly of all 4 extremities. Nails: distal onycholysis and beau’s lines in the first finger of the hands bilaterally. At the moment there is no evidence of mammary alterations.
Given the clinical findings, syndromic ectrodactyly of genetic origin was suspected and massively parallel sequencing of the individual clinical exome using NGS (Next Generation Sequencing) methodology was requested.
For the study the laboratory extracted and purified the patient’s DNA from the sample received, prepared a library of genome fragments; Selection of the regions under study by hybridization with probes in solution, which included the exons and adjacent intronic regions of the genes in the panel; Clonal amplification and sequencing of the selected regions on the illumina platform, HiSeq following the paired-ends strategy. GenSKits Reagents (IVD-CE). Sample coverage was 140.39X; bioinformatics study of the DNA sequence obtained, by comparison with the reference genomic sequence (GRCh38). Variant annotations those alterations presenting a read number ≥20X and a variant/reads ratio ≥0.2. GeneSystems platform (IDV-CE) [9]. Confirmation by Sanger sequencing of identified pathogenic or probably pathogenic changes. Variants of uncertain significance were not confirmed by this methodology. In silico predictive studies [10] of variants of uncertain significance using the Varsome program [11]. Analytical, interpretation and reporting practices adhering to current international standards.
The study reported a nucleotide variant c.739C>A in heterozygosity in exon 5/14 of the NM_003722.5 transcript of the TP63 gene. The p. His247Asn protein change is located in a conserved region. Confirmation was performed by Sanger sequencing.
The identified variant has not been reported in NCBI ClinVar, Medgen, HGMD, OMIM, 1000 Genomes Project, ExAc, LOVD3, Emsembl, gnomAD, ClinGen databases, but high-throughput bioinformatics algorithms predict a deleterious effect. In-Silico Predictors Meta scores report it as pathogenic PP3 classification: MetalR, MetaRNNN, BayesDel addAF, BayesDel non-AF, MetaSVM, REVEL, as do individual predictors: DEOGEN2, MutPred, EIGEN, EIGEN PC, FATHMM, M-CAP, PROVEAN, FATHMM-MKL, FATHMM-XF, LIST-S2, LRT, PrimateAI, SIFT, SIFT4G, BLOSUM, DANN, Mutation assessor, MutationTaster, MVP.
Pathogenic variants in this gene are associated with several phenotypes and medical conditions including ADULT syndrome with autosomal dominant inheritance mechanism [1].
Bioinformatics allows the management of large volumes of data that are used to verify information on populations previously studied with representative samples, optimizing the analysis of the information, however now the associated use of artificial intelligence confers greater reliability in the results, quality in the products and security therefore higher quality [13], which supports our approach and the use of these tools in order to perform a faster and more reliable genotype/phenotype correlation when facing cases like the one of our patient with initial suspicion of syndromic ectrodactyly of genetic origin.
Ectrodactyly is a rare congenital malformation of the extremities, in which there is a deep cleft in the central region of hands and/or feet, this disorder can occur in isolation or in conjunction with other manifestations constituting a syndromic presentation. It is described in 5 of the 8 clinical phenotypes associated with the heterozygous pathogenic variants of the TP63 gene, which is also expressed in epithelial and mesenchymal structures, hence its alterations also affect these tissues [7]. Its variants have also been associated with congenital ectodermal dysplasia, which are rare genetic disorders of the skin and appendages with hypodontia, hypotrichosis and hypohidrosis as main characteristics [12] including ADULT syndrome which presents both limb anomalies and previously mentioned findings of ectodermal dysplasia with anomalies, ungular, hair anomalies, hypo or oligodontia and ephelides which are normally marked; of these findings our patient had all of them except for the ephelides, which although they are considered a quite specific sign, it is not a constant finding in all cases because they can develop in early childhood, later or be absent without being a mandatory criterion for diagnosis [14]. We emphasize the importance of the initial approach to syndromic ectrodactyly because it was one of the first findings documented in our patient and even with prenatal ultrasound studies, in addition it is necessary to recognize that given the phenotypic heterogeneity, genetic studies are vital to make an adequate diagnosis reinforcing the concept of precision medicine generating an impact in the diagnosis. For this purpose, the use of high performance bioinformatics algorithms with the application of artificial intelligence have become fundamental allies that allow the analysis of big data in a shorter time, speeding up the analysis of the variants found and also the prediction of their effect in a reliable way.
ADULT syndrome (Acro-dermato-ungual-lacrimal-tooth syndrome), is related to pathogenic variants mainly in the transactivator domain of the TP63 gene, however, the variant reported in this case, is in the DNA binding domain a constant region and in our search it is not reported in NCBI databases, ClinVar, Medgen, OMIM, LOVD3, Emsembl, HGMD, Exac 1000 genomes, gnomAD, ClinGen, but different In-Silico Meta scores predictors report it as pathogenic PP3 classification, as well as the individual predictors described in the results [10].
The TP63 gene encodes a member of the p53 family of transcription factors, known as tumor protein p63. TP63 plays a crucial role in the development and maintenance of epithelial tissues, including skin, limbs and various organs [1]. It is involved in processes such as cell proliferation, differentiation and apoptosis. The p63 protein contains a transactivation domain, a DNA-binding domain, an oligomerization domain and a Sterile Alpha Motif (SAM) domain, which are essential for its function as a transcription factor.
This gene, also known as EEC3; Keratinocyte transcription factor KET; P73L; TP73L; Transformation-related protein 63; Tumor protein 63; Tumor protein p73-like; amplified in squamous cell carcinoma; p73H; tumor protein p53-competing protein; tumor protein p63; Chronic ulcerative stomatitis protein; KET; LMS; P63; TP53CP; TP53L; keratinocyte transcription factor KET; p53CP; p73L; tumor protein p63 deltaN isoform delta; tumor protein p63 deltaN isoform δ; tumor protein p73-like; B(p51A); B(p51B); CUSP; NBP; OFC8; P73H; RHS; SHFM4; TP63; chronic ulcerative stomatitis protein; p63; transformation-related protein 63; tumor protein 63; tumor protein p53-like; tumor protein p63 deltaN isoform Δ.Transcript NM_003722.4, ChromoChromosome 3q28. Domains: SAM, Start 541 End 607. Ammoniacids 680:
Identifier UP000005640 [11].
In accordance with various genomic intelligence platforms as mastermind [14], copilot [15] and VarChat [16] the functional domains of p53 family proteins include an N-terminal transac- tivation domain, a central DNA-binding domain and an oligo- merization domain. Alternative splicing of this gene and the use of alternative promoters results in multiple transcript variants encoding different isoforms that vary in their functional prop- erties. These isoforms function during skin development and maintenance, adult stem/progenitor cell regulation, heart de- velopment and premature aging. Some isoforms have been found to protect the germline by eliminating oocytes or testicu- lar germ cells that have suffered DNA damage [14-16].
These proteins play critical roles in cellular processes such as cell cycle regulation, DNA repair, and apoptpsis. Here are the key domains:
Transactivation Domain (AD)
Is essential for activating gene transcription. It interacts with other transcriptional machinery components and RNA poly- merase to initiate gene expression.
p53, p63, and p73 all share this domain. Interestingly, recent evidence indicates that all three proteins undergo N-terminal truncation, affecting the composition and activity of their ADs1.
Full-length p53 contains two tandem, independent ADs, while DNp53 (a dominant-negative form) has only one AD.
p63 and p73 also have ADs that transactivate p53 target genes1.
DNA-Binding Domain (DBD)
The DBD allows p53 family proteins to bind specifically to DNA sequences.
It is crucial for recognizing and binding to p53-responsive elements in target gene promoters.
p53, p63, and p73 all share significant similarity in their DBDs.
Tetramerization Domain (TD)
The TD facilitates the formation of tetramers (four protein subunits) for stable binding to DNA.
Tetramerization enhances the stability and DNA-binding ca- pacity of p53 family proteins.
Nuclear Localization and Export Signals (NLS and NES):
These signals regulate the transport of p53 family proteins between the nucleus andcytoplasm.
Proper localization is crucial for their functions.
Basic Domain (BD)
Present in p53 but not in p63 or p73.
Its specific role is not fully understood, but it likely con- tributes to protein-proteininteractions.
Sterile Alpha Motif (SAM) Domain
Present in some p63 and p73 isoforms but absent in p53.
Its function is not entirely clear, but it may be involved in pro- tein-protein interactions. In summary, while these proteins share common domains, they also exhibit distinct properties. Understanding their functional domains helps unravel their roles in various cellular processes, including development, maintenance, and protection against DNA damage [14-16].
The p. His247Asn variant is located within the DNA-binding domain of the p63 protein, which is highly conserved and is criti- cal in the protein’s ability to bind to specific DNA sequences and regulate target gene expression. The histidine at position 247 is a residue that may be involved in interactionswith the DNA backbone or in the structural integrity of the domain. Substitu- tion with asparagine potentially alters these interactions or the conformation of the DNA-binding domain, thus affecting the transcriptional regulatory activity of the p63 protein.
This change results in a codon-level missense variant involv- ing the substitution of a Cytosine (C) foran Adenine (A) at nucle- otide position 739 of the coding sequence.
The functional consequences of this variant depend on its impact on TP63 protein structure and function, and its effect on TP63-governed transcriptional networks.
Alpha Fold (Protein Structure Database) [17]; UniProt [18] Q9H3D4 Protein existence: evidence at protein level
Annotation score: 5/5. Acts as a sequence specific DNA bind- ing transcriptional activator or repressor.The isoforms contain a varying set of transactivation and auto-regulating transactiva- tion inhibiting domains thus showing an isoform specific activ- ity. Isoform 2 activates RIPK4 transcription. May berequired in conjunction with TP73/p73 for initiation of p53/TP53 depen- dent apoptosis in response togenotoxic insults and the presence of activated oncogenes. Involved in Notch signaling by probably inducing JAG1 and JAG2. Plays a role in the regulation of epi- thelial morphogenesis. The ratio of DeltaN-type and TA*-type isoforms may govern the maintenance of epithelial stem cell compartments and regulate the initiation of epithelial stratifica- tion from the undifferentiated embryonal ectoderm. Required for limb formation from the apical ectodermal ridge. Activates transcription of the p21 promoter [17,18].
It has molecular function: activator, developmental protein, DNA-binding.
Biological process: Apoptosis, Notch signaling pathway, Transcription, Transcriptionregulation
Ligand: Metal-binding, Zinc.
Pathway Commons: Q9H3D4.
Reactome: R-HSA-139915 Activation of PUMA and trans- location to mitochondria, R-HSA-5620971 Pyroptosis,R-HSA- 5628897TP53 Regulates Metabolic Genes, R-HSA-6803204TP53 Regulates Transcription of Genes Involved in Cytochrome C Re- lease, R-HSA-6803205TP53 Regulates Transcription of several additional cell death genes whose specific roles in p53-depen- dent apoptosis remain uncertain, -R-HSA-6803207TP53 Regu- lates Transcription of Caspase.
Activators and caspases
R-HSA-680323211TP53 Regulates Transcription of Death Re- ceptors and Ligands.
R-HSA-6804759Regulation of TP53 Activity through Associa- tion with Co-factors.
Proteomic databases: EPD Q9H3D4, MassIVE Q9H3D4, MaxQB Q9H3D4, PaxDb 9606- ENSP00000264731,PeptideAtlas Q9H3D4.
Expression tissue specificity: Widely expressed, notably in heart, kidney, placenta, prostate, skeletal muscle, testis and thymus, although the precise isoform varies according to tis- sue type. Progenitor cell layers of skin, breast, eye and pros- tate express high levels of DeltaN-type isoforms. Isoform 10 is predominantly expressed in skin squamous cell carcinomas, but not in normal skin tissues.
Interaction subunit binds: DNA as a homotetramer. Isoform composition of the tetramer may determine transactivation ac- tivity. Isoforms Alpha and Gamma interact with HIPK2. Interacts with SSRP1, leading to stimulate coactivator activity. Isoform 1 and isoform 2 interact with WWP1. Interacts with PDS5A. Iso- form 5 (via activation domain) interacts with NOC2L. Q9H3D4 has binaryinteractions with 17 proteins.
Protein-protein interaction databases
BioGRID 114181298; DIP interactors: DIP-29588N; ELM Q9H3D4; IntAct Q9H3D492; interactors MINTQ9H3D4; STRING 9606.ENSP00000264731.
Phylogenomic databases:
GeneTree: ENSGT00950000183153;
HOGENOM: CLU_019621_1_0_1; InParanoid: Q9H3D4; OMA: IRMQDSE; OrthoDB: 2902631 at 2759; PhylomeDB: Q9H3D4; TreeFam: TF106101; eggNOG: ENOG502QQ48.
Genome annotation databases:
Ensembl: ENST00000264731.8ENSP00000264731.3 ENSG00000073282.14 [Q9H3D4-1]; ENST00000320472.9EN SP00000317510.5ENSG00000073282.14 [Q9H3D4-7]; ENST 00000354600.10ENSP00000346614.5ENSG00000073282.14 [Q9H3D4-2]; ENST00000392460.7ENSP00000376253.3EN SG00000073282.14 [Q9H3D4-3]; ENST00000392461.7ENSP00 000376254.3ENSG00000073282.14 [Q9H3D4-8]; GeneID 8626; KEGG hsa:8626;
MANE-Select ENST00000264731.8ENSP00000264731.3NM_ 003722.5NP_003713.3;
UCSC uc003frx.3
This deleterious effect reported by the various in silico meth- ods, individual predictors and MetaScorepreviously described, along with the knowledge on functionality, biological bases, genomic and molecular annotation data, protein structure and function, functional, proteomic, experimental, pathways, gene expression, phylogenomic, ligand and cellular studies, and the use of artificial intelligence tools, allows the establish- ment of a genotype/phenotype correlation, gene expression, phylogenomics, ligands, cellular, use of artificial intelligence tools, allows establishing the genotype/phenotype correlation, constituting a new variant and given that the mechanism of in- heritance is autosomal dominant and there is no related family history, it is a de novo variant.
The identification and interpretation of new and de novo variants allows us to broaden and deepen our knowledge of genotypic and phenotypic heterogeneity, bringing us closer to a precision, personalized, predictive, preventive, participatory and proactive medicine with a view to being implemented at the population level.
In summary, we report the case of a patient with a variant in the TP63 gene not previously reported in literature or databas- es, with no related family history in non-consanguineous parents associated with ADULT syndrome, a condition with autosomal dominant inheritance mechanism; thus constituting a new and de novo variant.
ADULT syndrome is a rare genetic disease with an autoso- mal dominant inheritance pattern, secondary to heterozygous pathogenic variants in the TP63 gene, represented by limb anomalies, ectodermal dysplasia, associated with alterations of the lacrimal duct, mammary hypoplasia and ephelides among others, we report the case of a patient with a characteristic phe- notype with genetic study that documents the new nucleotide variant c.739C>A, in the DNA binding domain; in a patientwith no family history of genetic diseases, parents and sibling without phenotypic alterations, or otherpathological history, so it is also a de novo variant involved in the genesis of the condition re- ported in the patient.
After the research carried out until May 2024, no report was found in the different databases for the identified variant. How- ever, the different high-performance bioinformatics algorithms, individual predictors and MetaScore, along with the knowl- edge about functionality, biological bases, genomic annotation data, molecular, protein structure and function, functional, proteomic, experimental, pathways, gene expression, phyloge- nomic, ligand, cellular studies, use of artificial intelligence tools which predict a deleterious effect on the variant and according to Richards, et al. Standards and guidelines for the interpreta- tion of sequence variants. American college of medical genetics and genomics, association for molecular pathology, ClinGen this variant is classified as PS3, PM2, PS4, PM6, PP3, allows to classify the variant as probably pathogenic and to establish the genotype/phenotype correlation [19].
Given this, it is concluded that we are facing a complex and uncommon case with a new and denovo variant in a gene as- sociated with multiple clinical conditions (according to Human Phenotype Ontology -HPO) [20] but in our patient with manifes- tations on one of them the ADULT syndrome, achieving through the use of bioinformatics algorithms and applied artificial intel- ligence tools, establishing the genotype/phenotype correlation, reinforcing the concept of precision medicine, in order to reach an early and accurate diagnosis which is fundamental for an ad- equate therapeutic approach with the prevention of complica- tions, genetic counseling and scientific contribution [8,21].
